Case study: 1K PBMC ATAC
In this case study, we will explain how to process 10X ATACseq by using the following test dataset: 1k Peripheral Blood Mononuclear Cells (PBMCs) from a Healthy Donor (Next GEM v1.1)
Learning objectives
Download input data
Here we will download the 1k Peripheral Blood Mononuclear Cells (PBMCs) from a Healthy Donor (Next GEM v1.1) and the human reference genome for Cell Ranger ATAC:
Download ATACseq data
mkdir tests/PBMC_1K_ATAC && cd tests/PBMC_1K_ATAC
# Input fastq files
wget https://cf.10xgenomics.com/samples/cell-atac/2.0.0/atac_pbmc_1k_nextgem/atac_pbmc_1k_nextgem_fastqs.tar
# Human reference genome
wget "https://cf.10xgenomics.com/supp/cell-arc/refdata-cellranger-arc-GRCh38-2024-A.tar.gz"
Extract files
tar -xvf atac_pbmc_1k_nextgem_fastqs.tar
tar -xvf refdata-cellranger-arc-GRCh38-2024-A.tar.gz
Set up the input files
Initialize a config file:
pipeline_config.yaml
Run the following command to generate a config file with all available ATAC parameters:
sc-preprocess init-config --modality atac --output pipeline_config.yaml
This writes every parameter with its default value and inline comments. Fields marked # REQUIRED must be filled in before running. Optional steps (doublet_detection, demultiplexing) are included but set to enabled: false — set them to true to activate them. For demultiplexing, both method blocks (demuxalot and vireo) are always printed — only the one matching method: is used; the other is ignored.
Make a
libraries.tsv
This file tells the pipeline where to find your FASTQ files. It has four tab-separated columns: batch, capture, sample, and fastqs.
echo -e "batch\tcapture\tsample\tfastqs" > libraries.tsv
echo -e "1\tL001\tatac_pbmc_1k_nextgem\t$(realpath atac_pbmc_1k_nextgem_fastqs)" >> libraries.tsv
Column |
Description |
|---|---|
|
Batch identifier (e.g. |
|
Lane/capture identifier (e.g. |
|
Sample name passed to |
|
Absolute path to the directory containing FASTQ files |
📌 Note: The
fastqscolumn must be an absolute path —cellranger-atacwill fail with relative paths. The$(realpath ...)call above handles this automatically.
Update
pipeline_config.yaml
Fill in any other required paths (e.g. path to libaries.tsv which points to your input fasta files), parameters, as well as enabled/disable and steps so the workflow fits your single-cell preprocessing needs. Today, we will be running cellranger-atac count to map the single-cell RNAseq data and Scrublet to identify doublets.
For this case study, fill out the pipeline_config.yaml with these value and adjust paths to file where necessary:
project_name: 1K_PBMC_ATAC_PROCESSED # REQUIRED
output_dir: 1K_PBMC_ATAC_PROCESSED # REQUIRED
resources:
mem_gb: 32 # default memory; individual steps override this with their own mem_gb
tmpdir: "" # temp directory for large file operations
directories_suffix: none # suffix appended to output directory names; "none" to disable
cellranger_atac:
enabled: true
reference: /path/to/reference # REQUIRED
libraries: libraries.tsv # REQUIRED: TSV with columns: batch, capture, sample, fastqs
chemistry: auto # options: auto, ARC-v1
normalize: none # options: none, depth
threads: 10
mem_gb: 64
runtime_minutes: 720
cluster-mode: # cellranger-atac cluster-mode: see https://www.10xgenomics.com/support/software/cell-ranger-atac/latest/advanced/cluster-mode
enabled: false # set true to submit Cell Ranger ATAC jobs via a cluster scheduler
jobmode: slurm # slurm | lsf | sge | /path/to/custom.template
mempercore: null # SLURM users: leave null — memory is requested directly via --mem=__MRO_MEM_GB__G
maxjobs: 64 # max concurrent cluster subjobs
jobinterval: null # delay between submissions in ms; increase if cluster rate-limits
# cellranger_atac_aggr — runtime scales with number of captures; 4+ captures can take 5–7h
aggr:
threads: 32
mem_gb: 64
runtime_minutes: 480
# create_atac_anndata (includes fragment sort — can take 10–30 min on real data)
anndata:
threads: 16
mem_gb: 32
runtime_minutes: 120
# aggregate_atac_batch
batch_aggregation:
threads: 16
mem_gb: 32
runtime_minutes: 120
doublet_detection:
enabled: True
method: scrublet # only supported method
threads: 1
mem_gb: 16
scrublet:
filter_cells_min_genes: 100
filter_genes_min_cells: 3
expected_doublet_rate: 0.06
min_gene_variability_pctl: 85.0
n_prin_comps: 30
sim_doublet_ratio: 2.0
threshold: null # doublet score cutoff; auto-determined if null
n_neighbors: null # KNN neighbors; auto-set to 0.5*sqrt(n_obs) if null
random_state: 0
demultiplexing:
enabled: false
method: vireo # options: demuxalot, vireo — only the selected method is used
demuxalot: # genotype-based; GEX only
vcf: /path/to/genotypes.vcf # REQUIRED
genome_names: /path/to/genome_names.txt # REQUIRED
refine: false # run genotype refinement step
celltag: CB
umitag: UB
vireo: # SNP-based; works with all modalities
donors: 4 # REQUIRED: number of donors
cellsnp:
vcf: /path/to/variants.vcf # REQUIRED
threads: 4
min_maf: 0.0
min_count: 1
umi_tag: Auto
cell_tag: CB
gzip: true
Run the tool
Dry-run
Before running the workflow it’s best practice to run a dry-run - a Snakemake command that will test the workflow without executing the underlying rules and print out it’s gameplan for every job in the workflow. The most informative part for us is the Job stats section which we highlight below. Job stats counts how many times individual Rules will be run and acts as a fantastic sanity check prior to executing the workflow. For example, if you have three multiome captures, then the Rule cellranger_atac_count should be run three times. In this case study, we only have one capture so all Rules are executed once.
# Read about this command
sc-preprocess run -h
# Dry run
$ sc-preprocess run --config-file pipeline_config.yaml --cores 1 --dry-run
[INFO] Config validated. Enabled steps: cellranger_atac, doublet_detection
[INFO] Running Snakemake with command: snakemake --snakefile ~/github/sc-preprocess/sc_preprocess/workflows/main.smk --configfile pipeline_config.yaml --cores 1 --use-conda --dry-run
[INFO] ============================================================
[INFO] Single-Cell Preprocessing Pipeline
[INFO] ============================================================
[INFO] Project: 1K_PBMC_ATAC_PROCESSED
[INFO] Output directory: 1K_PBMC_ATAC_PROCESSED
[INFO] Enabled steps: cellranger_atac, doublet_detection
[INFO] ============================================================
[INFO] libraries.tsv file format is valid.
[INFO] libraries.tsv file format is valid.
[INFO] Cell Ranger ATAC: Found 1 sample(s) across 1 batch(es)
[INFO] Cell Ranger ATAC: Found 1 sample(s) across 1 batch(es)
[INFO] Batch aggregation: Found 1 ATAC batch(es)
[INFO] Batch aggregation: Found 1 ATAC batch(es)
[INFO] Doublet Detection: Using scrublet method
[INFO] Doublet Detection: Using scrublet method
host: midway3-login1.rcc.local
Building DAG of jobs...
Job stats:
job count
--------------------- -------
cellranger_atac_count 1
cellranger_atac_aggr 1
create_atac_anndata 1
aggregate_atac_batch 1
run_scrublet 1
enrich_atac_metadata 1
all 1
total 7
...
Visualize the workflow with a DAG file
Our favorite way to visualize a dry-run of a workflow is to examine the DAG file. This image represents the network of jobs and dependencies found in the dry-run of the workflow. Each node is a job and each arrow represents a dependent rule.
📌 Note: If the rules are circles then the rule has not been run yet, however, if the rules are bordered with dotted lines then it’s been completed. This distinction is valuable when examining an incomplete workflow.
sc-preprocess run --config-file pipeline_config.yaml --cores 1 --dag | dot -Tpng > dag_atac_1k.png
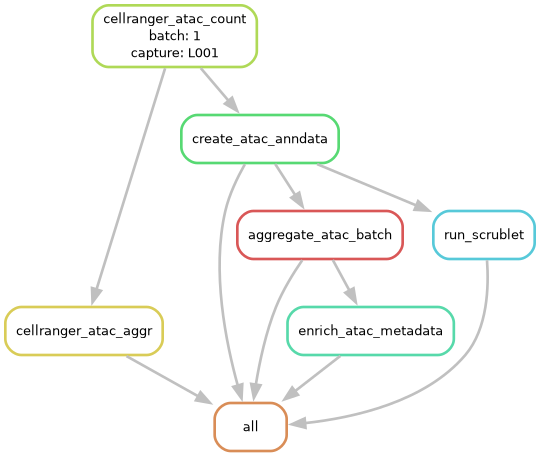
ATAC Pipeline DAG — DAG file showing all rules and their dependencies.
Rule descriptions
Here we will break down the meaning of each rule so you can keep track of what’s going on.
cellranger_atac_count: Runs the command cellranger-atac count per capture, aligning ATAC reads to the reference genome and producing a peak-barcode matrix.
create_atac_anndata: Converts Cell Ranger ATAC output to a per-capture AnnData object (
.h5ad), adding traceability metadata (batch_id,capture_id,cell_id). Before importing fragments into SnapATAC2, the pipeline sortsfragments.tsv.gzby barcode and caches the result asfragments.sorted_by_barcode.tsv.gznext to the original file. This sort is the most resource-intensive step in the pipeline: on real data a fragments file is typically 2–5 GB and sorting it can take 10–30 minutes and require 16–32 GB of RAM. Useanndata.mem_gbandanndata.runtime_minutesin the config to size the job accordingly. The sorted file is cached — if you re-run the pipeline it will be reused automatically.cellranger_atac_aggr: Runs cellranger-atac aggr which aggregates all per-capture Cell Ranger ATAC outputs within a batch into a single normalized count matrix.
aggregate_atac_batch: Merges all per-capture AnnData objects into a single batch-level
.h5adfile, verifyingcell_iduniqueness across captures.run_scrublet: Runs Scrublet doublet detection on each per-capture AnnData object, adding doublet scores and predictions to cell metadata.
enrich_atac_metadata: Joins all downstream preprocessing metadata from doublet detection into the batch-level AnnData object.
all: Final Snakemake rule that collects all expected outputs to ensure the full workflow is completed.
Local Execution
# Local execution
sc-preprocess run --config-file pipeline_config.yaml --cores 1
The parameter --cores is a Snakemake argument that defines the maximum number of CPU cores Snakemake can use at any given time across all running tasks locally. You can read about it here.
Snakemake arguments
We added the parameter --snakemake-args to send arguments straight to Snakemake!
For example, a popular Snakemake argument is --keep-going, where Snakemake will continue running jobs even if one fails. Please note that it MUST be the last argument in the command. Here is what it looks like in practice:
sc-preprocess run --config-file pipeline_config.yaml \
--cores 1 \
--dry-run \
--snakemake-args --keep-going
Another useful argument is --forcerun, which forces Snakemake to re-execute a specific rule and all rules that depend on it — without re-running expensive upstream steps like Cell Ranger. This is handy when you update a script and only want to reprocess from that point forward:
sc-preprocess run --config-file pipeline_config.yaml \
--cores 1 \
--snakemake-args "--forcerun create_atac_anndata aggregate_atac_batch"
📌 Note: You can pass multiple rule names to
--forcerun. Snakemake will automatically re-run all downstream rules that depend on the forced rules.
Run jobs in parallel!
Snakemake shines when jobs can be run in parallel across multiple nodes in a cloud or HPC environment. Here, we will discuss how to leverage SLURM but Snakemake can easily be plugged in other HPC environments.You can read more about the Snakemake SLURM configuration file here.
📌 Note: Replace the BASH variables
SLURM_ACCOUNTandSLURM_PARTITIONwith your SLURM appropriate setting before running the script below.
Here is a quick way to make the SLURM HPC profile:
# Set your SLURM account and partition
SLURM_ACCOUNT="" # <- replace with your account
SLURM_PARTITION="" # <- replace with your partition
mkdir -p HPC_profiles
cat > HPC_profiles/config.yaml << EOF
executor: slurm
jobs: 10
default-resources:
- slurm_account=${SLURM_ACCOUNT}
- slurm_partition=${SLURM_PARTITION}
- runtime=720
retries: 2
latency-wait: 60
printshellcmds: true
keep-going: true
rerun-incomplete: true
EOF
Now you can run it like this:
# HPC execution - `--cores all` tells Snakemake to use the `threads` assigned to each rule.
sc-preprocess run --config-file pipeline_config.yaml \
--cores all \
--snakemake-args --profile HPC_profiles --keep-going
Parallel computing example
Here are two snippets from the files, HPC_profiles/config.yaml and pipeline_config.yaml, and how they would manifest if launched on an HPC
HPC_profiles/config.yaml
executor: slurm
jobs: 10
default-resources:
- slurm_account={SLURM-ACCOUNT}
- slurm_partition={SLURM-PARTITION}
- runtime=720
retries: 2
latency-wait: 60
printshellcmds: true
keep-going: true
rerun-incomplete: true
jobs= how many SLURM jobs Snakemake submits simultaneously to any node. Each Snakemake rule execution is one job.
pipeline_config.yaml
cellranger_atac:
enabled: true
reference: /path/to/reference/refdata-cellranger-arc-GRCh38-2020-A-2.0.0 # REQUIRED
libraries: libraries.tsv # REQUIRED: TSV with columns: batch, capture, sample, fastqs
chemistry: auto # options: auto, ARC-v1
normalize: none # options: none, depth
threads: 10
mem_gb: 64
runtime_minutes: 720
cluster-mode: # cellranger-atac cluster-mode: see https://www.10xgenomics.com/support/software/cell-ranger-atac/latest/advanced/cluster-mode
enabled: false # set true to submit Cell Ranger ATAC jobs via a cluster scheduler
jobmode: slurm # slurm | lsf | sge | /path/to/custom.template
mempercore: null # SLURM users: leave null — memory is requested directly via --mem=__MRO_MEM_GB__G
maxjobs: 64 # max concurrent cluster subjobs
jobinterval: null # delay between submissions in ms; increase if cluster rate-limits
# cellranger_atac_aggr — runtime scales with number of captures; 4+ captures can take 5–7h
aggr:
threads: 32
mem_gb: 64
runtime_minutes: 480
# create_atac_anndata (includes fragment sort — can take 10–30 min on real data)
anndata:
threads: 16
mem_gb: 32
runtime_minutes: 120
# aggregate_atac_batch
batch_aggregation:
threads: 16
mem_gb: 32
runtime_minutes: 120
Running sc-processes with the files above will look like this across your HPC:
jobs: 10
└─ job 1: cellranger_atac_count (batch1_L001) → 1 node, 10 CPUs, 64 GB, 720 min
└─ job 2: create_atac_anndata (batch1_L001) → 1 node, 16 CPUs, 32 GB, 120 min ← includes fragment sort
└─ job 3: aggregate_atac_batch (batch1) → 1 node, 16 CPUs, 32 GB, 120 min
└─ ...up to 10 running at once
Cell Ranger cluster-mode
By default, cellranger-atac count runs all of its internal pipeline stages on the same node as the Snakemake job. On large datasets this can be slow because Cell Ranger ATAC is limited to the cores allocated to that single SLURM job.
Cluster-mode lets Cell Ranger ATAC submit each of its internal pipeline stages as independent SLURM jobs, parallelizing the work across your cluster. You can read about how to set it up on the 10X website here: https://www.10xgenomics.com/support/software/cell-ranger-atac/latest/advanced/cluster-mode.
Here are some descriptions of the cluster-mode parameters in pipeline_config.yaml:
Field |
Description |
|---|---|
|
|
|
|
|
SLURM users: leave |
|
Maximum number of Cell Ranger ATAC subjobs submitted at once |
|
Milliseconds between job submissions (optional, default is fine) |
To enable, add a cluster-mode: block with enabled: true to cellranger_atac in your config:
cellranger_atac:
enabled: true
reference: /path/to/cellranger-atac/reference # REQUIRED
libraries: /path/to/libraries.tsv # REQUIRED: TSV with columns: batch, capture, sample, fastqs
chemistry: auto # options: auto, ARC-v1
normalize: none # options: none, depth
threads: 10
mem_gb: 64
runtime_minutes: 720
cluster-mode: # cellranger-atac cluster-mode: see https://www.10xgenomics.com/support/software/cell-ranger-atac/latest/advanced/cluster-mode
enabled: false # set true to submit Cell Ranger ATAC jobs via a cluster scheduler
jobmode: slurm # slurm | lsf | sge | /path/to/custom.template
mempercore: null # SLURM users: leave null — memory is requested directly via --mem=__MRO_MEM_GB__G
maxjobs: 64 # max concurrent cluster subjobs
jobinterval: null # delay between submissions in ms; increase if cluster rate-limits
# cellranger_atac_aggr — runtime scales with number of captures; 4+ captures can take 5–7h
aggr:
threads: 32
mem_gb: 64
runtime_minutes: 480
# create_atac_anndata (includes fragment sort — can take 10–30 min on real data)
anndata:
threads: 16
mem_gb: 32
runtime_minutes: 120
# aggregate_atac_batch
batch_aggregation:
threads: 16
mem_gb: 32
runtime_minutes: 120
With cluster-mode enabled in the files above, running sc-processes will look like this across your HPC:
jobs: 10
└─ job 1: cellranger_atac_count (batch1_L001) → 1 node, 10 CPUs, 64 GB ← launcher
└─ CR sub-job 1 → separate SLURM job (managed by Cell Ranger, not Snakemake)
└─ CR sub-job 2 → separate SLURM job
└─ ... up to maxjobs: 64
└─ job 2: create_atac_anndata (batch1_L001) → 1 node, 16 CPUs, 32 GB, 120 min ← includes fragment sort
You can confirm cluster mode is active by checking the log file — each pipeline stage will show (run:slurm) instead of (run:local):
2026-04-13 13:20:25 [runtime] (run:slurm) ID.1_L001.SC_ATAC_COUNTER_CS.WRITE_GENE_INDEX.fork0.chnk0.main
2026-04-13 13:20:37 [runtime] (run:slurm) ID.1_L001.SC_ATAC_COUNTER_CS.DETECT_CHEMISTRY.fork0.chnk0.main
📌 Note: When you configure cluster-mode template be aware that you can launch jobs to different partitions and nodes than the
sc-preprocessjobs which are controlled by theHPC_profile.
Resuming a killed run: If the job is killed mid-run, Cell Ranger ATAC’s Martian runtime saves checkpoints after each completed stage. The pipeline automatically removes the _lock file left by the killed process, so resubmitting the Snakemake job will resume from the last completed stage rather than starting over.
Interpreting STDOUT
After starting the program you should see an output that looks like this, let’s break it down:
$ sc-preprocess run --config-file pipeline_config.yaml \
> --cores all \
> --snakemake-args --profile HPC_profiles --keep-going
[INFO] Config validated. Enabled steps: cellranger_atac, doublet_detection
[INFO] Running Snakemake with command: snakemake --snakefile ~/github/sc-preprocess/sc_preprocess/workflows/main.smk --configfile pipeline_config.yaml --cores all --use-conda --profile HPC_profiles --keep-going
Using profile HPC_profiles for setting default command line arguments.
[INFO] ============================================================
[INFO] Single-Cell Preprocessing Pipeline
[INFO] ============================================================
[INFO] Project: 1K_PBMC_ATAC_PROCESSED
[INFO] Output directory: 1K_PBMC_ATAC_PROCESSED
[INFO] Enabled steps: cellranger_atac, doublet_detection
[INFO] ============================================================
[INFO] libraries.tsv file format is valid.
[INFO] libraries.tsv file format is valid.
[INFO] Cell Ranger ATAC: Found 1 sample(s) across 1 batch(es)
[INFO] Cell Ranger ATAC: Found 1 sample(s) across 1 batch(es)
[INFO] Batch aggregation: Found 1 ATAC batch(es)
[INFO] Batch aggregation: Found 1 ATAC batch(es)
[INFO] Doublet Detection: Using scrublet method
[INFO] Doublet Detection: Using scrublet method
host: midway3-login1.rcc.local
Building DAG of jobs...
SLURM run ID: fe7a5e53-100a-411c-9d66-de1ec86b684c
MinJobAge 120s (>= 120s). 'squeue' should work reliably for status queries.
Using shell: /usr/bin/bash
Provided remote nodes: 10
Job stats:
job count
--------------------- -------
cellranger_atac_count 1
cellranger_atac_aggr 1
create_atac_anndata 1
aggregate_atac_batch 1
run_scrublet 1
enrich_atac_metadata 1
all 1
total 7
Select jobs to execute...
Execute 1 jobs...
Messages from this tool will always be prefaced in brackets e.g. [INFO], [WARNING], [ERROR].
The first [INFO] prints the preprocessing steps enabled in the config file. In this tutorial, we enabled Cell Ranger ATAC to process the 1k PBMCs ATAC data and Doublet detection with Scrublet:
[INFO] Config validated. Enabled steps: cellranger_atac, doublet_detection
Next, we print the Snakemake command running under the hood for convenient debugging. The --snakefile path will reflect where sc-preprocess is installed in your environment — this is expected and you don’t need to use this path directly.
[INFO] Running Snakemake with command: snakemake --snakefile /path/to/sc_preprocess/workflows/main.smk --configfile pipeline_config.yaml --cores all --use-conda --profile HPC_profiles
After that, we print some more [INFO] about the run:
[INFO] ============================================================
[INFO] Single-Cell Preprocessing Pipeline
[INFO] ============================================================
[INFO] Project: 1K_PBMC_ATAC_PROCESSED
[INFO] Output directory: 1K_PBMC_ATAC_PROCESSED
[INFO] Enabled steps: cellranger_atac, doublet_detection
[INFO] ============================================================
Finally, we print [INFO] from every job so you can fact check your workflow i.e. are these the number of batches and samples you were expecting to preprocess?
[INFO] libraries.tsv file format is valid.
[INFO] Cell Ranger ATAC: Found 1 sample(s) across 1 batch(es)
[INFO] Batch aggregation: Found 1 ATAC batch(es)
[INFO] Doublet Detection: Using scrublet method
📌 Note: You may see some messages printed twice. This is expected — Snakemake evaluates the config in two passes during DAG construction.
Everything else are messages directly from Snakemake running the workflow you configured! If you are new to Snakemake please take some time to orient yourself: https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html
Log file paths for every Rule will be printed in the Snakemake stdout like this:
log: 1K_PBMC_ATAC_PROCESSED/00_LOGS/1_L001_atac_count.log
For example, you could explore the log for that Cell Ranger ATAC job by printing the log file like this:
$ cat 1K_PBMC_ATAC_PROCESSED/00_LOGS/1_L001_atac_count.log
Martian Runtime - v4.0.7
Serving UI at http://midway3-0323.rcc.local:34101?auth=upepGFywWPU8K2GA4BD29lPWcYSjdgQbAakoEbQTQS0
Running preflight checks (please wait)...
Checking FASTQ folder...
Checking reference...
Checking reference_path (/path/to/refdata-cellranger-arc-GRCh38-2024-A) on midway3-0323.rcc.local...
Checking optional arguments...
...
2026-04-12 16:27:23 [runtime] (chunks_complete) ID.1_L001.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ATAC_MATRIX_COMPUTER.MARK_ATAC_DUPLICATES
2026-04-12 16:27:23 [runtime] (run:local) ID.1_L001.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ATAC_MATRIX_COMPUTER.MARK_ATAC_DUPLICATES.fork0.join
Examine the output directory structure
After successfully completing the workflow, you should see this resulting directory structure. Let’s break it down:
$ tree -L 2 1K_PBMC_ATAC_PROCESSED/
1K_PBMC_ATAC_PROCESSED/
├── 00_LOGS
│ ├── 1_L001_atac_count.done
│ ├── 1_L001_atac_count.log
│ ├── 1_L001_atac_anndata.done
│ ├── 1_L001_atac_anndata.log
│ ├── 1_atac_aggr.done
│ ├── 1_atac_aggr.log
│ ├── 1_atac_batch_aggregation.done
│ ├── 1_atac_batch_aggregation.log
│ ├── 1_atac_enrichment.done
│ ├── 1_atac_enrichment.log
│ ├── 1_L001_scrublet.done
│ └── 1_L001_scrublet.log
├── 01_CELLRANGERATAC_COUNT
│ └── 1_L001
├── 02_CELLRANGERATAC_AGGR
│ └── 1_aggregation.csv
├── 03_ANNDATA
│ └── 1_L001.h5ad
├── 04_BATCH_OBJECTS
│ └── 1_atac.h5ad
├── 06_DOUBLET_DETECTION
│ └── 1_L001_scrublet.tsv.gz
└── 07_FINAL
├── 1_atac.h5ad
├── 1_atac_obs_summary.tsv.gz
└── 1_atac_obs.tsv.gz
00_LOGS/
This directory contains all the .log and .done files created throughout the workflow and are organized by Batch_Capture_modality_rule. The .log files will contain any STDOUT printed from every step of the workflow. This allows you to dive in and interrogate any step of your single-cell preprocessing.
A quick way to find errors if you are debugging the workflow is to run:
grep -R "error" 1K_PBMC_ATAC_PROCESSED/00_LOGS
The .done files are an internal checklist to keep track of a subset of rules that finished (don’t worry about it unless you are a developer and want to contribute to the code base).
01_CELLRANGERATAC_COUNT/
Here you will find all of the Cell Ranger ATAC count outputs for each individual capture.
02_CELLRANGERATAC_AGGR/
This will be the aggregated count matrices across batches. In this tutorial there is only one capture so you won’t find any processed data here.
03_ANNDATA/
Here you will find an AnnData object for every capture.
04_BATCH_OBJECTS/
Batch-level AnnData object created by merging all per-capture objects from 03_ANNDATA/. This is the aggregated, pre-metadata-enriched object — all cells from all captures in the batch are present, and cell_id uniqueness is verified. It does not yet contain doublet scores or demultiplexing results.
06_DOUBLET_DETECTION/
Doublet detection outputs from Scrublet.
07_FINAL/
The final enriched AnnData object with all preprocessing metadata joined in, ready for downstream analysis.
1_atac.h5ad
1_atac_obs_summary.tsv.gz
1_atac_obs.tsv.gz
Load the output for downstream analysis
Examine barcode metadata
Now that you have successfully preprocessed the dataset 1k PBMCs ATAC we will show a few examples of how you can immediately start analyzing your data!
Check out a summary of the workflow and barcode metadata with these files:
1_atac_obs.tsv.gz
$ python -c "import pandas as pd; df = pd.read_csv('1K_PBMC_ATAC_PROCESSED/07_FINAL/1_atac_obs.tsv.gz', sep='\t'); print(df)"
cell_id n_fragment frac_dup frac_mito batch_id capture_id ... total_counts doublet_scrublet_scrublet_score doublet_scrublet_scrublet_predicted_doublet
0 1_L001_AAACGAATCGCATAAC-1 16220 0.621161 0.0 1 L001 ... 0.0 NaN NaN
1 1_L001_AAACGAATCTGTGTGA-1 7256 0.636455 0.0 1 L001 ... 0.0 NaN NaN
2 1_L001_AAACTCGAGAGGAACA-1 17324 0.592951 0.0 1 L001 ... 0.0 NaN NaN
...
[1016 rows x 14 columns]
1_atac_obs_summary.tsv.gz
$ python -c "import pandas as pd; df = pd.read_csv('1K_PBMC_ATAC_PROCESSED/07_FINAL/1_atac_obs_summary.tsv.gz', sep='\t'); print(df)"
batch_id n_cells median_fragments median_peaks
0 1 1016 0.0 0.0
SnapATAC2
📌 Note: A companion Jupyter notebook for loading the output and generating QC visualizations is available at
notebooks/PBMC_1k_ATAC_analysis.ipynb.
The pipeline produces two objects for ATAC analysis:
03_ANNDATA/1_L001_snap.h5ad— SnapATAC2-native per-capture object. Stores raw fragment data inobsm['fragment_paired'], which is required for fragment-level analyses like TSS enrichment scoring and fragment size distribution plots.07_FINAL/1_atac.h5ad— Final enriched scanpy AnnData. Has the cells × peaks matrix, all QC metrics, traceability metadata, and doublet scores — but no raw fragment data.
Load the SnapATAC2-native object for fragment-level QC:
import snapatac2 as snap
adata_snap = snap.read_dataset("1K_PBMC_ATAC_PROCESSED/03_ANNDATA/1_L001_snap.h5ad")
# Fragment size distribution (nucleosomal banding pattern)
fig = snap.pl.frag_size_distr(adata_snap, show=False)
fig.update_yaxes(type="log")
fig.show()
# TSS enrichment score
snap.metrics.tsse(adata_snap, snap.genome.hg38)
snap.pl.tsse(adata_snap)
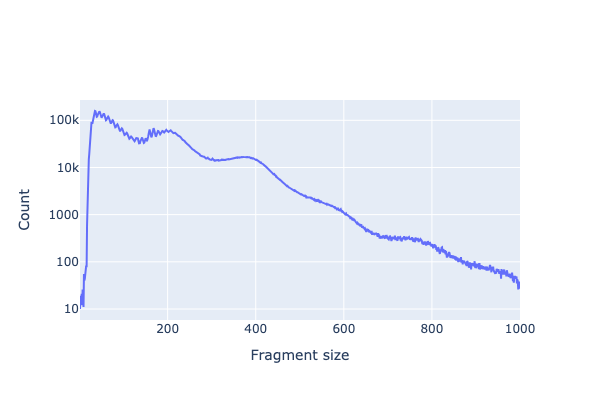
Fragment size distribution — nucleosomal banding pattern from 1K PBMC ATAC data.
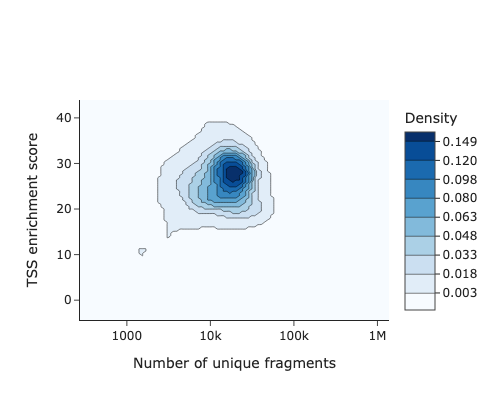
TSS enrichment — accessibility signal at transcription start sites.
For common questions about re-running steps, .done file tracking, and cluster-mode lock files, see the FAQ.